Our Disease Focus

At Rocket, we are in pursuit of gene therapy cures for patients living with rare and devastating genetic diseases.
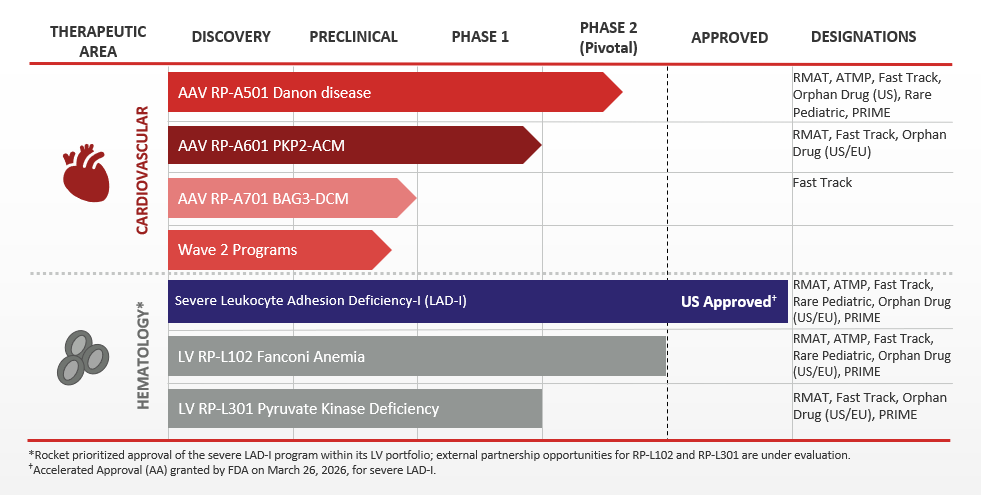
Our Pipeline
Rocket’s pipeline is comprised of first-in-class gene therapies that incorporate either AAV or LV approaches to gene therapy.
Cardiovascular
Danon disease is a rare genetic disorder that is characterized by severe and primarily hypertrophic cardiomyopathy. Skeletal muscle weakness and mild cognitive impairment are also common. The causative mutation has been identified in the gene encoding for lysosome-associated membrane protein (LAMP2), and in particular the LAMP2B version of the gene which is primarily expressed in heart, skeletal muscle and brain tissue. Affected cells cannot efficiently digest and recycle protein and other materials, leading to dysfunction in heart, muscle and brain cells. Male patients who don’t receive a heart transplant often pass away during adolescence or early adulthood from progressive heart failure.
RP-A501 is an investigational gene therapy consisting of a recombinant adeno-associated serotype 9 (AAV9) capsid containing a functional version of the human LAMP2B transgene (AAV9.LAMP2B) which is administered as a single intravenous (IV) infusion. RP-A501 has the potential to restore or stabilize cardiac function in patients with Danon disease.
Plakophilin-2 related arrhythmogenic cardiomyopathy (PKP2-ACM), also known as ARVC or ARVD, is a devastating, inherited heart disease caused by mutations in the PKP2 gene and associated with life-threatening arrhythmias, cardiac structural abnormalities, and sudden cardiac death. Patients living with PKP2-ACM have an urgent unmet medical need, as current medical, implantable cardioverter defibrillator (ICD), and ablation therapies do not consistently prevent disease progression or arrhythmia recurrence, are associated with significant morbidity including inappropriate shocks and device and procedure-related complications, and do not address the underlying pathophysiology or genetic mutation.
RP-A601 is an investigational gene therapy consisting of a recombinant adeno-associated serotype rh74 (AAVrh74) capsid containing a functional version of the human PKP2 transgene (AAVrh74.PKP2) which is administered as a single intravenous (IV) infusion. RP-A601 has the potential to restore or stabilize cardiac function in patients with PKP2-ACM.
BAG3-associated dilated cardiomyopathy (BAG3-DCM) is a rare genetically driven form of heart failure caused by mutations in the BAG3 gene. The BAG3 protein is expressed predominantly in the heart, where it plays a role in multiple key cellular functions of the heart. Loss of BAG3 leads to an accumulation of misfolded and damaged proteins, which can impair the heart’s ability to contract, potentially leading to impaired cardiac function, heart failure, and even premature death.
RP-A701 is an investigational gene therapy consisting of a recombinant adeno-associated serotype rh74 (AAVrh74) capsid. Because BAG3 plays a role in multiple critical myocardial cell functions, we believe RP-A701 has the potential to restore or stabilize cardiac function in patients with BAG3-DCM.
Hematology
Leukocyte adhesion deficiency type I (LAD-I) is an ultra-rare genetic pediatric disease caused by mutations in the ITGB2 gene encoding for CD18, a key protein that is expressed along CD11 integrins to facilitate leukocyte adhesion to the blood vessel wall and migration to tissues to confine and clear infections and orchestrate wound repair. Infants with severe LAD-I suffer from recurrent, life-threatening bacterial, and fungal infections that respond poorly to antimicrobials and require frequent hospitalizations.
Learn more about a product available for severe LAD-I here.
Fanconi Anemia (FA) is a rare genetic disorder affecting DNA repair and characterized by bone marrow failure (BMF), cancer predisposition, and congenital malformations. Approximately two-thirds of FA cases are caused by genetic defects in the FANCA gene, which results in the FA subtype known as FA Complementation Group A (FA-A). FA patients may develop severe bone marrow failure (very low blood counts), cancers of the blood or other cancers. Currently, donor-mediated bone marrow transplants are the most frequently utilized therapy for FA.
RP-L102 is an investigational gene therapy containing autologous (patient-derived) hematopoietic stem cells (HSCs) that have been genetically modified with a lentiviral vector to contain a functional copy of the FANCA gene. If given early in life, RP-L102 gene therapy has the potential to correct bone marrow cells and stabilize blood counts, avoiding progression to severe bone marrow failure.
Pyruvate Kinase Deficiency (PKD) is a rare genetic blood disorder characterized by excessive rupture of red blood cells (RBCs), resulting in frequent, chronic anemia that can lead to life-threatening complications for patients with the severe form of the disease. Patients with PKD frequently have anemia, chronic fatigue, yellowing of the skin and eyes (jaundice) and enlarged spleens.
RP-L301 is an investigational gene therapy containing autologous (patient-derived) hematopoietic stem cells (HSCs) that have been genetically modified with a lentiviral vector to contain a functional copy of the PKLR gene. RP-L301 has the potential to correct multiple disease elements associated with PKD.
Wave 2 Progams
We are using our successful disease-first approach to advance multiple undisclosed candidates to potential clinical development.